Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone


State Key Laboratory of Estuarine and Coastal Research, East China Normal University, Shanghai 200062, China
*
Author to whom correspondence should be addressed.
Water 2019, 11(6), 1261; https://doi.org/10.3390/w11061261
Submission received: 24 April 2019
/
Revised: 30 May 2019
/
Accepted: 31 May 2019
/
Published: 17 June 2019
(This article belongs to the Section Water Quality and Contamination)
Abstract
:Nutrients and organic pollutants transported by submarine groundwater discharge (SGD) play a significant role in controlling water quality, and can lead to the concerned deleterious effects on marine ecosystems. Subterranean estuaries are complicated habitats of diverse microbial communities that mediate different biogeochemical processes. However, there is less information on how microorganisms mediate biogeochemical cycles in the submarine groundwater system. In this study, we investigated the changes in bacterial and archaeal assemblages from two size fractions (0.2–0.45 μm and >0.45 μm) in the submarine groundwater of Qinzhou Bay, China. Phylogenetic analysis showed that Bathyarchaeota was dominant in archaeal communities in the >0.45 μm size fraction, but was seldom in the 0.2–0.45 μm fraction. The co-occurrence of sequences belonging to Bathyarchaeota and Methanosaeta was found in the >0.45 μm size fraction. Since a gene encoding acetate kinase of Bathyarchaeota is involved in acetate production, and acetate is also a necessary growth factor for Methanosaeta, the acetate produced by Bathyarchaeota can provide food or energy sources for Methanosaeta in this very >0.45 μm size fraction. The most abundant bacterial sequences in the >0.45 μm size fraction was closely related to biomineral iron-oxidizing Gallionella spp., whereas the dominant bacterial sequences in the 0.2–0.45 μm fraction were affiliated with Limnohabitans spp., which can utilize dissolved organic matter as an important source of growth substrates. Notably, approximately 10% of the bacterial sequences in both of the two size fractions belonged to Novosphingobium spp., which plays an important role in the degradation of pollutants, especially aromatic compounds. Furthermore, the predictive functional profiling also revealed that the pathways involved in the degradation of aromatic compounds by both bacteria and archaea were identified. The presence of nutrients or pollutants in our study site provides different substrates for the growth of the specific microbial groups; in turn, these microbes may help to deplete pollutants to the ocean through submarine groundwater. We suggest that these specific microbial groups could be potential candidates for effective in situ bioremediation of groundwater ecosystems.
1. Introduction
Submarine groundwater discharge (SGD) is defined as all the water flow on continental margins from the seabed to the coastal ocean, without regard to their fluid composition or driving force [1]. Compared with surface water, submarine groundwater has a different geochemistry, and where these waters mix prior to discharging, they form reactive zones known as the subterranean estuary [2]. The composition of SGD differs from that predicted by simple mixing, because biogeochemical reactions in the aquifer modify its chemistry [3]. SGD fluxes of nutrients, metals, carbon and organic pollutants (e.g., pesticides, polychlorinated biphenyls (PCBs), polycyclic aromatic hydrocarbons (PAHs) and phthalates) are significant pathways from land to sea [3,4,5], and have been recognized to have negative effects, such as red tide outbreaks [6], eutrophication [7], and hypoxia [8], on marine ecological environments [9]. Therefore, SGD and its associated chemical substances are considered key parts of coastal management [10].
Qinzhou Bay is located in the south of Qinzhou City, Guangxi province, and is an industrial region greatly affected by industrial activities [11,12] (Figure 1). Recently, the natural ecosystem structure and marine ecological environment of this region have been changed because of industrialization and urbanization, such as the construction of large-scale petroleum refineries and pulp and paper mills [13,14]. Our recent work also revealed that significant SGD-derived nutrient and carbon fluxes exceeded river inputs in this region [11,12]. In this study, the sampling site is located in the Gaoshatou Village of Qinzhou Bay (Figure 1). The contamination of aromatic compounds often occurs near the industrial zone. For example, PCBs are widely used in industry, and their concentrations (ng g−1 dry wt) range from 1.62 to 62.6, with a mean of 9.87 in Qinzhou Bay [13]. The station with the highest PCBs concentration is very close to the compact district of the aforementioned petrochemical plants. However, there are no reports on the microbial compositions in the submarine groundwater of Qinzhou Bay, especially in different size fractions.
Suspended particulate matter (SPM) (>0.45 μm) is one of the main forms of nutrients, that is, carbon and organic pollutants which are transferred from land to the marine environment [15,16]. Composed of nutrients, organic micro-pollutants and heavy metals, SPM can affect material exchange and biogeochemical processes in coastal systems [15,17]. A number of studies demonstrated that microbial community composition, abundance and activity vary between the water and SPM in coastal surface water (e.g., [18,19,20]).
However, there are few reports on microorganisms from different size fractions in SGD, especially with reference to microorganisms attached to SPM. SGD is now globally recognized as one of the most important and central processes of land-ocean interaction in coastal zones [3]. Previous studies suggested that SGD is a significant pathway for fecal indicator bacteria to enter coastal waters in submarine groundwater contaminated by sewage, through activities such as waste treatment and disposal (e.g., [21]). In our previous work, bacterial diversity and distribution were investigated in SGD along the coastal zone of the Yellow Sea, and we found some potential key bacterial groups, such as Comamonas spp., that may be excellent candidates for the bioremediation of natural pollutants in the SGD [22].
Microorganisms play an important role in mediating biochemical reactions within groundwater systems [21,23]. In this study, 16S rRNA gene-based MiSeq Illumina sequencing approach and PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) were used to analyze the bacterial and archaeal community compositions, and predict functional profiling between the 0.2–0.45 μm and >0.45 μm fractions (i.e., SPM) in the submarine groundwater of Qinzhou Bay, China, respectively. The objective of this work was (1) to explore the specific microbial groups in these two different size fractions, which could play an important ecological role in biogeochemical cycles in SGD to coastal zones. (2) To identify potential microbial candidates for in situ bioremediation/biodegradation in coastal groundwater ecosystems, and then provide valuable information for coastal environmental management.
2. Materials and Methods
2.1. Sample Collection and Measurements of Chemical Parameters
The samples were collected during March 2017, from an abandoned well near an industry zone in the Gaoshatou Village of Qinzhou Bay (Figure 1). Well water near the coasts is typical submarine groundwater, and can result in significant impacts on marine ecosystems [1,9,22]. In this study, submarine groundwater was collected by using an organic glass hydrophore [11,12]. The salinity and temperature were measured by a YSI-EC300A conductivity meter (YSI Inc., Yellow Springs, OH, USA) in the field. 222Rn samples were collected in glass bottles using the overflow method [11,12], and were immediately analyzed using the RAD7 detector (Durridge Co. Inc., Billerica, MA, USA). Nutrients, dissolved inorganic carbon (DIC), and dissolved organic carbon (DOC) samples, were collected using polyethylene bottles filtered by 0.45 µm cellulose acetate filters, and stored in the dark to inhibit microbial growth [12,24]. The nutrient concentrations (NO2−, NO3−, NH4+, PO43− and SiO32−) were then analyzed using an autoanalyzer (Model: Skalar SANplus system, Skalar Analytical B.V., Breda, The Netherlands) [24]. The concentration of dissolved inorganic nitrogen (DIN) was determined as the sum of NO2−, NO3− and NH4+. The dissolved inorganic phosphorus (DIP) and dissolved inorganic silicon (DSi) represent the concentrations of PO43− and SiO32−, respectively. The DIC and DOC were analyzed using a TOC Analyzer (TOC-VCPH, Shimadzu Co. Ltd., Kyoto, Japan) [12].
To collect 0.2–0.45 μm and >0.45 μm size fractions, ten liter samples of submarine groundwater were first filtered through 0.45 µm pore size cellulose acetate filters, then filtered through 0.2 µm pore size polycarbonate filters (Nuclepore Track-Etched Membrane, Whatman Inc. Clifton, NJ, USA). Since the heterogenic geological matrix and high dynamics of aquifers can change the biogeochemical parameters of submarine groundwater [11,25], to eliminate accidental errors caused by heterogeneity and dynamics, quadruplicate submarine groundwater samples were collected size fractions. Both the 0.45 µm pore size cellulose acetate filters and 0.2 µm pore size polycarbonate filters were placed in sterile 1.5 mL microcentrifuge tubes, and immediately stored at −20 °C onsite, then samples were subsequently stored at −80 °C until further analysis. The blank filter treated with sterile water of the same volume as in the environmental sample was used as a negative control.
2.2. DNA Extraction, PCR Amplification and Illumina MiSeq Sequencing
Total environmental DNA was extracted from quadruplicate filters (four 0.45 µm filters and four 0.2 µm filters) using the MoBio PowerWater® DNA Isolation Kit (MOBIO Laboratories, Carlsbad, CA, USA) according to the manufacturer’s instructions. Both the concentration and purity of the DNA were assessed by spectrophotometry (NanoDrop ND1000 spectrophotometer, Thermo Fisher Scientific, Wilmington, DE, USA). The extracted DNA was stored at −20 °C until further analysis. The results of the DNA purity and the concentration for each sample were showed in Table S1.
Minimum numbers of polymerase chain reaction (PCR) cycles were performed, and three independent PCR mixtures were pooled for each sample to decrease PCR bias. The bacterial 16S rRNA genes containing V4–V5 hypervariable regions were amplified using primers 515F (5′-GTGCCAGCMGCCGCGG-3′) and 907R (5′-CCGTCAATTCMTTTRAGTTT-3′) [26], and the following amplification conditions: 2 min at 95 °C; followed by 25 cycles of 95 °C for 30 s, 30 s at 55 °C, and 30 s at 72 °C; and an extension at 72 °C for 5 min. The archaeal 16S rRNA genes were amplified using primers 524F10extF (5′-TGYCAGCCGCCGCGGTAA-3′) and Arch958RmodR (5′-YCCGGCGTTGAVTCCAATT-3′) [27], using the following amplification conditions: 3 min at 95 °C; followed by 35 cycles of 30 s at 95 °C, 30 s at 55 °C, and 45 s at 72 °C; and a final extension at 72 °C for 10 min.
For Illumina MiSeq sequencing, PCR products were purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s protocol, and then quantified by QuantiFluorTM-ST (Promega, Madison, WI, USA). Reaction mixtures were pooled in equimolar ratios, and paired-end reads were generated on an Illumina MiSeq PE250 (Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China). Negative control did not produce a detectable PCR fragment for either bacterial or archaeal 16S rRNA gene amplification.
2.3. Sequence Data Processing, OTU Clustering, and Taxonomic Assignment
Raw Illumina FASTQ files were demultiplexed, quality filtered and analyzed using Quantitative Insights into Microbial Ecology (QIIME) (version 1.17) (An open-source software pipeline available at http://qiime.sourceforge.net/) [28] with criteria described previously [29]. Operational taxonomic units (OTUs, 97% similarity cutoff) were clustered using UPARSE (version 7.1) (An open-source software pipeline available at http://drive5.com/uparse/). Chimeric sequences were screened using UCHIME. The abundances of OTUs from each sample was determined by OTU cluster. The number of reads from each sample was assigned to each out, and an “OTU table” was generated using the usearch_global command. To obtain the taxonomic information of each OTU corresponding species, the Ribosomal Database Project (RDP) Classifier (http://rdp.cme.msu.edu/) was used for taxonomic analysis of representative OTU sequences. The community composition of each sample was calculated at the genus level. Sequence data were entered into the NCBI Sequence Read Archive under BioProject ID PRJNA515097 (bacteria) and PRJNA515099 (archaea).
2.4. Phylogenetic Analyses
The sequences of the representative OTUs in this study were analyzed in GenBank by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to obtain reference sequences. The sequences of representative OTUs and selected reference sequences from the database were aligned using Clustal W. Then the phylogenetic trees were generated in MEGA6 using the neighbor-joining method with a bootstrap test of 1000 replicates and a maximum composite likelihood model [30].
2.5. Statistical Analyses
Alpha diversity metrics and coverage were calculated by the Mothur program [31]. As a visualization technique, principal-coordinate analyses (PCoA) using Bray-Curtis distance was performed to show if distinct separations in bacterial or archaeal community structures were present between the 0.2–0.45 μm and >0.45 μm size fractions. PERmutational Multivariate ANalysis Of VAriance (PERMANOVA) based on this Bray-Curtis distance was constructed in R package vegan. PERMANOVA calculation was performed using raw relative abundance data.
2.6. Predictive Functional Profiling
Based on the 16S rRNA gene sequences, PICRUSt was used to the predictive functional profiling of microbial communities [32]. First, the abundance of each OTU was normalized by the 16S rRNA gene copy number predictions. The abundance of Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology was then calculated according to the greengenes ID of each OTU. Here, the greengenes ID is obtained from the Greengenes database (http://greengenes.lbl.gov/). The final output table of gene family counts was summarized as pathway-level categories.
3. Results
3.1. Site Description and Environmental Characteristics
The descriptions of the sampling site and environmental chemical parameters of submarine groundwater in this study are summarized in Table 1. The study site is close to the industrial zone of Qinzhou City, China. The temperature and salinity of the submarine groundwater were 17.2 °C and 0.5, respectively. The activity of 222Rn in the studied well water (4140 Bq m−3) was much higher than that of 222Rn in the seawater (127 Bq m−3) of Qinzhou Bay. Choosing a representative submarine groundwater end-member is always a key and difficult problem in SGD studies [11,12,25,33]. This is because representative submarine groundwater end-member must take into account the salinity, 222Rn (an excellent SGD tracer) and the distance from the coastal line [11,12,25]. The values of salinity and 222Rn in this studied well, as well as its position, indicated that the studied well water is a representative submarine groundwater end member. The DIC concentration of the submarine groundwater was 1.3 mmol L−1 and the DOC concentration was 1.0 mmol L−1. The concentrations (μmol L−1) of DIN, DIP and DSi in the studied submarine groundwater were 10.4, 0.31 and 166, respectively.
3.2. Bacterial and Archaeal Diversity
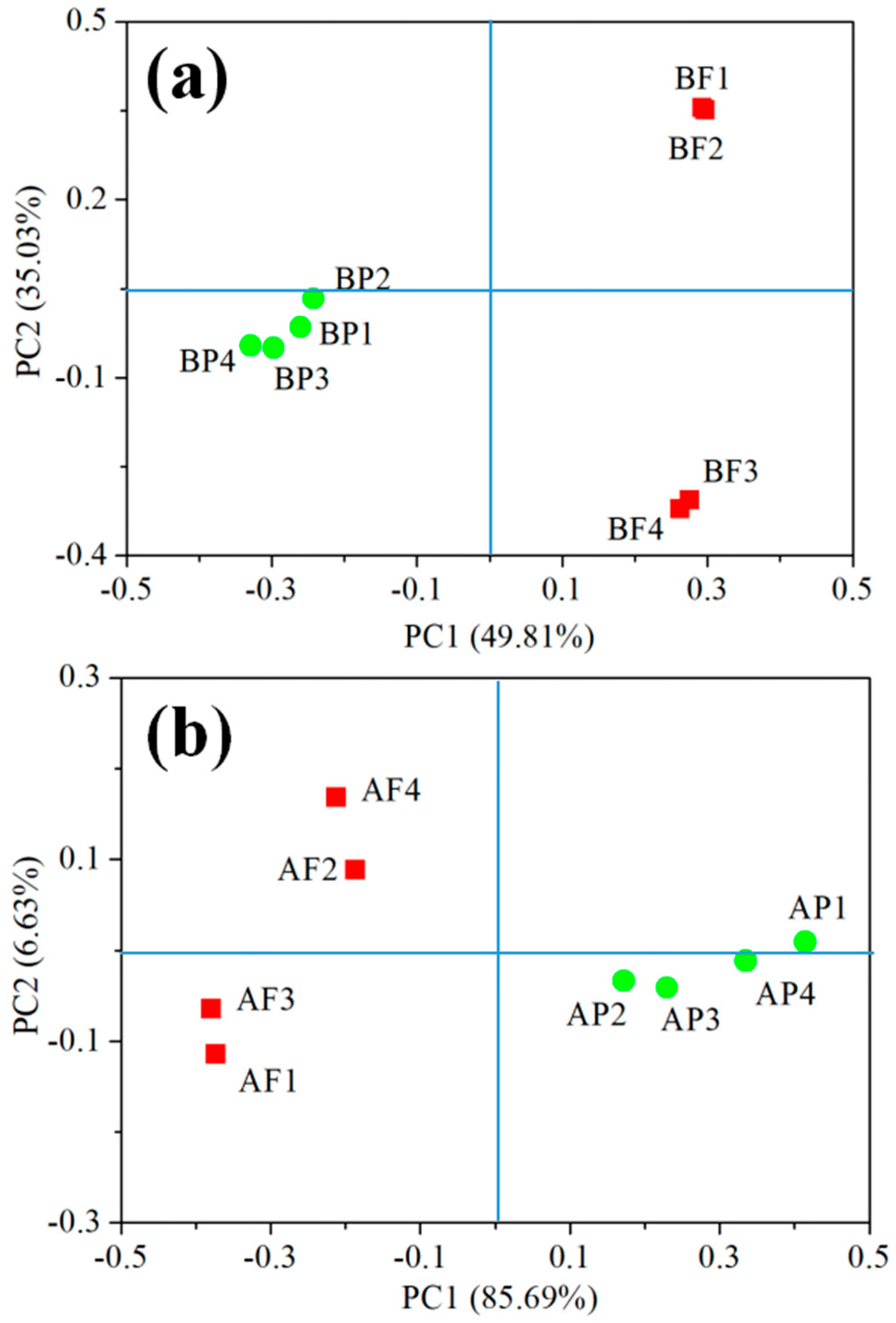
To obtain insights into the microbial diversity of the submarine groundwater in Qinzhou Bay, we obtained a total of 240,429 high-quality bacterial V4-V5 Illumina sequences and 345,993 high-quality archaeal V4-V5 Illumina sequences in two size fractions. There were 22,776 bacterial reads and 24,760 archaeal reads per sample after subsampling (all samples were randomly resampled down to the smallest sample size). According to the 97% similarity cutoff, there were 1113 bacterial OTUs and 619 archaeal OTUs in the complete OTU data set. Among these OTUs, 756 common bacterial OTUs and 451 common archaeal OTUs were found in the two size fractions. Good’s coverage was 99.1–99.9% for all samples. PCoA analysis showed that the bacterial community in the 0.2–0.45 μm fraction was clearly separated from that in the >0.45 μm fraction along the first axis, explaining 49.81% of the variation. Similarly, the archaeal community in the 0.2–0.45 μm fraction was clearly separated from that in the >0.45 μm fraction along the first axis explaining 85.69% of variation (Figure 2).
PCoA analysis also showed that the spatial pattern of the bacterial community in the two size fractions was opposite to the archaeal community, which may indicate that the spatial niche partitioning of bacterial and archaeal communities was different in the two size fractions of submarine groundwater. However, we found that quadruplicate submarine groundwater samples of bacteria and archaea in the 0.2–0.45 μm fraction were also separated, which may be attributed to the heterogenic geological matrix and high dynamics of aquifers [11,25]. In addition, the R2 and p values were calculated using the PERMANOVA approach, which showed that significant differences were found in both bacteria (PERMANOVA, n = 4, R2 = 0.51, p < 0.05) and archaea (PERMANOVA, n = 4, R2 = 0.78, p < 0.05) between the >0.45 μm size fraction and the 0.2–0.45 μm fraction (Table 2).
3.3. Bacterial and Archaeal Distribution
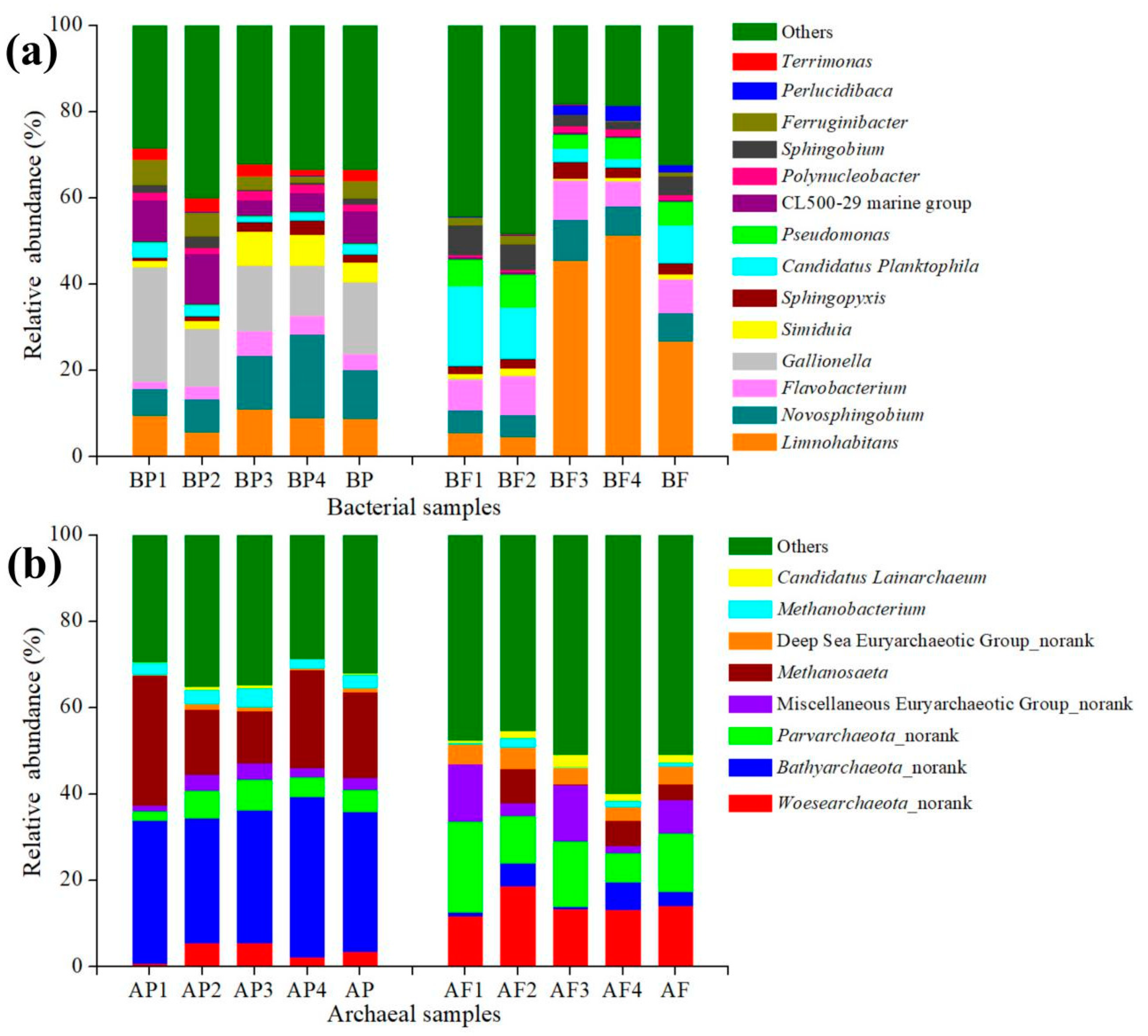
Taxonomic distributions show that there were some differences in the proportion of Illumina sequences between the >0.45 μm size fraction and the 0.2–0.45 μm fraction, respectively (Figure 3). Each percentile value in the parenthesis in the text below is the mean value of quadruplicate analyses. Limnohabitans and Gallionella were the most abundant genera in the 0.2–0.45 μm fraction (26.6%) and the >0.45 μm size fraction (16.7%) in bacterial samples, respectively. However, Limnohabitans existed in both size fractions, but Gallionella only existed in the >0.45 μm size fraction. The reason may be that Gallionella need to obtain energy for growth from Fe(II) oxidation and facilitate the precipitation of Fe(III) oxyhydroxides [34]. The second dominant groups were Candidatus Planktophila in the 0.2–0.45 μm fraction (8.8%) and Novosphingobium in the >0.45 μm size fraction (11.5%) in bacterial samples. Bathyarchaeota (32.5%) was the most abundant genus in the >0.45 μm size fraction archaeal sample and Methanosaeta (19.9%) formed the second dominant group in the >0.45 μm size fraction archaeal sample. However, Bathyarchaeota (3.3%) was seldom detected in the 0.2–0.45 μm size fraction. In addition, a large number of archaeal sequences (i.e., others: 51.1%) were remotely related to the reported environmental sequences in the database in the 0.2–0.45 μm size fraction, showing the archaeal communities in the 0.2–0.45 μm size fraction had more diversity.
3.4. Phylogenetic Analyses
3.4.1. Main Bacterial Groups in Two Size Fractions
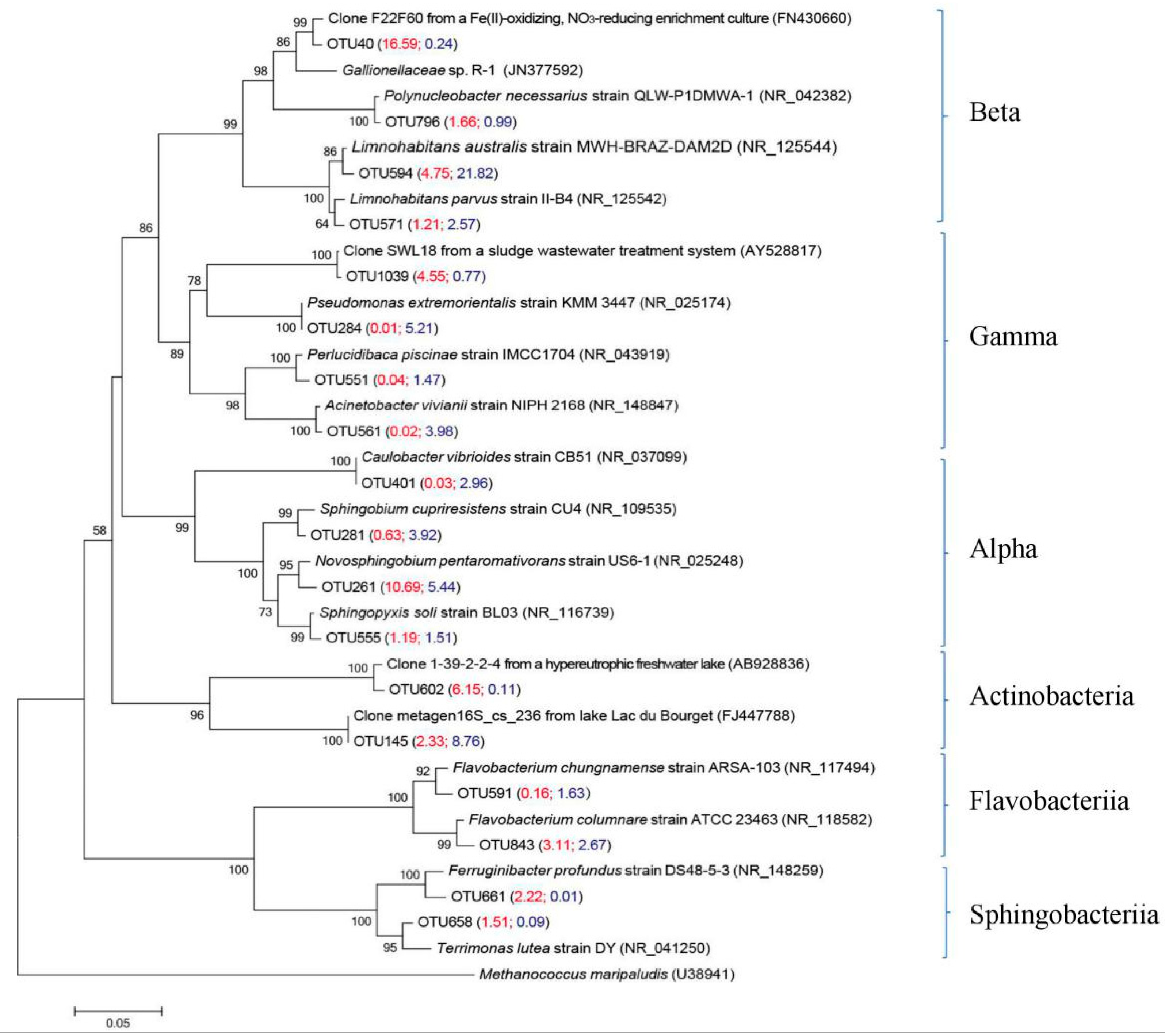
A bacterial analysis detected OTUs related to Betaproteobacteria, Gammaproteobacteria, Alphaproteobacteria, Actinobacteria, Flavobacteria and Sphingobacteria within the >0.45 μm size fraction and the 0.2–0.45 μm fraction (Figure 4). As shown in Table 3, the betaproteobacterial sequences of OTU 594 were the most abundant genus in the 0.2–0.45 μm fraction (21.8%), and were also found in the >0.45 μm size fraction (4.7%) and exhibited 99.2% similarity with the Limnohabitans australis strain MWH-BRAZ-DAM2D (NR_125544) [35]. Sequences of OTU 571 were obtained from the >0.45 μm size fraction (1.2%) and the 0.2–0.45 μm fraction (2.6%), and showed 98.9% similarity with the Limnohabitans parvus strain II-B4 (NR_125542) [36]. The betaproteobacterial sequences of OTU 40 were the most abundant genus in the >0.45 μm size fraction (16.6%), but were few in the 0.2–0.45 μm fraction (0.2%); this OTU had 98.9% similarity with the clone F22F60 from a Fe(II)-oxidizing, nitrate-reducing enrichment culture (FN430660) [37]. OTU 1039 was found in the >0.45 μm size fraction (4.5%), but there were very few in the 0.2–0.45 μm fraction (0.8%); this OTU was phylogenetically associated (99.7% similarity) with the gammaproteobacterial clone SWL18 from a sludge wastewater treatment system (AY528817) [38]. The alphaproteobacterial sequences of OTU 261 and OTU 555 were found in the >0.45 μm size fraction (10.7% for OTU 261 and 1.2% for OTU 555) and the 0.2–0.45 μm fraction (5.4% for OTU 261 and 1.5% for OTU 555). OTU 261 had 98.4% similarity with the Novosphingobium pentaromativorans strain US6-1 (NR_025248), which is an aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment [39], and OTU 555 had 99.2% similarity with the Sphingopyxis soli strain BL03 (NR_116739) [40]. Two OTUs displayed relatively high identities with representatives within the class Flavobacteria. OTU 843, representing 3.1% and 2.7% of the >0.45 μm size fraction and 0.2–0.45 μm fraction, respectively, had 98.7% similarity with the Flavobacterium chungnamense strain ARSA-103 (NR_117494) [41]. OTU 591 comprised 0.1% and 1.6% sequences in the >0.45 μm size fraction and the 0.2–0.45 μm fraction, respectively, and had 98.4% similarity with the Flavobacterium columnare strain ATCC 23463 (NR_118582) [42].
3.4.2. Main Archaeal Groups in Two Size Fractions
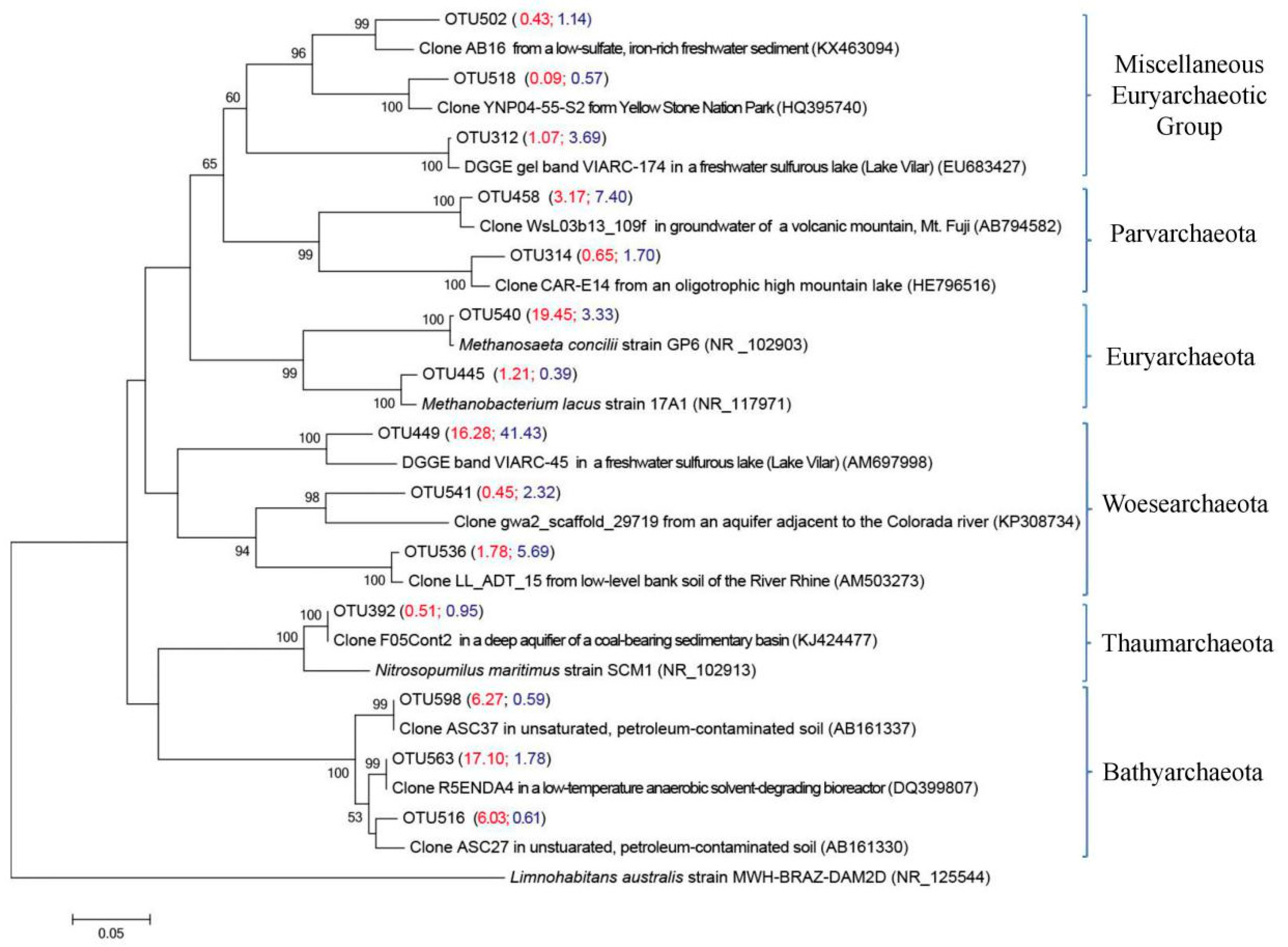
In the archaeal analysis, OTUs related to Bathyarchaeota, Euryarchaeota, Parvarchaeota, and Woesearchaeota were detected within the >0.45 μm size fraction and the 0.2–0.45 μm fraction (Figure 5). As shown in Table 4, phylogenetic analysis showed that three representative OTUs (OTU 563, 516 and 598) belonging to the phylum Bathyarchaeota were dominant in archaeal communities in the >0.45 μm size fraction (29.4%), but were seldom detected in the 0.2–0.45 μm fraction (3.0%). OTU 563 exhibited 100% similarity with clone R5ENDA4 from low-temperature anaerobic solvent-degrading bioreactors (DQ399807) [43]. OTU 516 showed 100% similarity with clone ASC27 in unsaturated, petroleum-contaminated soil (AB161330) [44], and OTU 598 had 100% similarity with clone ASC37 in unsaturated, petroleum-contaminated soil (AB161337) [44]. Euryarchaeotal OTU 540 was found in the >0.45 μm size fraction (19.5%), but was seldom detected in the 0.2–0.45 μm fraction (3.3%); this OTU gave a 99.6% match to the acetoclastic methanoarchaeon Methanosaeta concilii strain GP6 (NR_102903) [45]. Within the Miscellaneous Euryarchaeotic Group (MEG), OTU 312 was found in the >0.45 μm size fraction (1.1%) and 0.2–0.45 μm fraction (3.7%) and had 99.2% similarity with DGGE (Denaturing Gradient Gel Electrophoresis) gel band VIARC-174 from the water column of Lake Vilar (EU683427) [46]. Phylum Parvarchaeota was found in the >0.45 μm size fraction (3.2%) and the 0.2–0.45 μm fraction (7.4%). Representative OTU 458 had 98.4% similarity with the clone WsL03b13_109f from groundwater of a volcanic mountain, Mt. Fuji (AB794582) (Unpublished). The sequences related to phylum Woesearchaeota including OTU 536 and OTU 541 were found in the >0.45 μm size fraction (1.8% for OTU 536 and 0.5% for OTU 541) and the 0.2–0.45 μm fraction (5.7% for OTU 536 and 2.3% for OTU 541). OTU 536 had 98.8% similarity with clone LL_ADT_15 from low-level bank soil of the River Rhine (AM503273) [47], and OTU 541 had only 87.0% similarity with clone gwa2_scaffold_29719 from an aquifer adjacent to Colorado River (KP308734) [48]. Another important finding in this study is that the unidentified sequences of OTU 449 were abundantly found in the >0.45 μm size fraction (16.3%) and the 0.2–0.45 μm fraction (41.4%). This OTU had 92.5% similarity with VIARC-45 (AM697998) retrieved from the water column of Lake Vilar. VIARC-45 was grouped within Deep-Sea Hydrothermal Vent Euryarchaeotal Group 6 (DHVG-6) [46].
3.5. Distinct Bacterial and Archaeal Communities in Two Size Fractions
In this study, Gallionella spp. and Limnohabitans spp. constituted the major group in the >0.45 μm size fraction and the 0.2–0.45 μm fraction, respectively. Meanwhile, phylogenetic analysis showed that the dominant archaeal phylum in the >0.45 μm size fraction was Bathyarchaeota, which was seldom detected in the 0.2–0.45 μm size fraction. In most cases, 0.45 µm pore size membrane filters are commonly used to collect SPM samples [17], especially for geochemists. Therefore, to analyze the bacterial and archaeal communities attached to SPM, we used 0.45 µm pore size cellulose acetate membranes to collect the samples in the >0.45 μm size fraction. We found that Bathyarchaeota was dominant in archaeal communities in the >0.45 μm size fraction, but was seldom detected in the 0.2–0.45 μm size fraction, indicating that Bathyarchaeota were preferentially attached to the SPM with a particle size greater than 0.45 µm.
4. Discussion
4.1. The Ecological Niches of Bathyarchaeota in Submarine Groundwater Systems
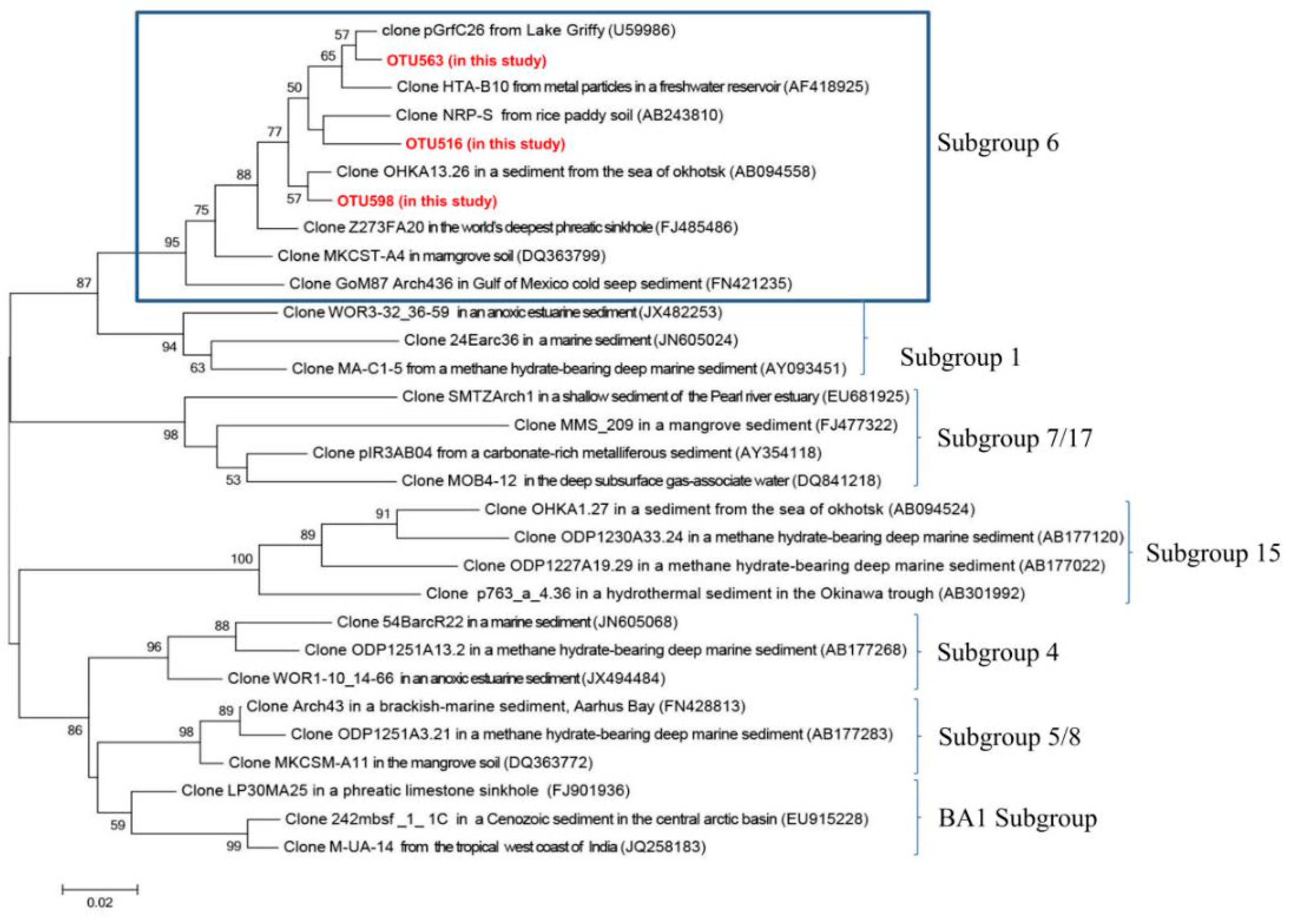
Phylogenetic analysis showed that the dominant archaeal group in the >0.45 μm size fraction was Bathyarchaeota. This Bathyarchaeota, which was formerly known as the Miscellaneous Crenarchaeotal Group (MCG), dominates marine subsurface archaeal communities [49,50] and has not yet been successfully cultured in the laboratory due to the slow growth rate of these microbes [51]. Recently, four genomic bins (MCG-6, MCG-1, MCG-7/17 and MCG-15) were found to belong to different Bathyarchaeota subgroups 6, 1, 7/17 and 15 [52]. Bathyarchaeota subgroup 6 has been proposed to have the ability to hydrolyze extracellular plant-derived carbohydrates, and degrade detritus proteins by metabolic predictions [52]. Meanwhile, all four subgroups have been shown to possess genes encoding enzymes involved in acetate production and the reductive acetyl-CoA pathway, suggesting that four subgroups of Bathyarchaeota are organo-heterotrophic and autotrophic acetogens, and these subgroups also have a potential metabolic pathway for dissimilatory nitrite reduction to ammonium [52]. In this study, we found that three dominant bathyarchaeotal OTUs (OTU 563, 516 and 598) were affiliated with our Bathyarchaeota subgroup 6 (Figure 6), showing similar ecological potentials to those of this subgroup 6. Based upon the estimation of chemical rates, Bathyarchaeota have been identified as some of the most active microbial groups in the deep marine biosphere [53]. Some studies have found that Bathyarchaeota not only has metabolic pathways to degrade complex organic compounds such as aromatic compounds, cellulose, chitin and proteins, but also can use hydrogen and CO2 to produce acetate [50,52,54,55]. Metabolic reconstruction based on genomic bins assembled from the metagenome found that the acetate produced by acetogenic Bathyarchaeota is an important electron carrier, and may be consumed by heterotrophic bacteria and acetoclastic methanogens, where the acetogenic Bathyarchaeota are key players in carbon cycling and ecosystems in marine sediments [55]. However, to our knowledge, there are few reports on the Bathyarchaeota in the submarine groundwater systems. Our results suggest that Bathyarchaeota are not only active and key players in carbon cycling and ecosystems in the deep marine biosphere, but also significant participants in carbon metabolism in submarine groundwater systems.
In addition, the BA1 and BA2 genomes of Bathyarchaeota contain divergent homologs of the genes necessary for methane metabolism obtained from metagenomic analysis [51]. Bathyarchaeota may play an important ecological role via a potential symbiotic association with Methanosaeta [56]. Acetate is an important organic substrate for the growth and methane production of acetoclastic Methanosaeta [57]. Meanwhile, a gene encoding acetate kinase of Bathyarchaeota could also be involved in acetate production [55,56]. In this study, the co-occurrence of sequences belonging to Bathyarchaeota and the genus Methanosaeta was found in the >0.45 μm size fraction, indicating possible acetate production by members of Bathyarchaeota in SPM. Since the concentrations of organic compounds such as PAHs in SPM were very high in comparison with those in water in a coastal ecosystem [58], SPM could provide a source of nutrients for Bathyarchaeota. The metabolic produced by Bathyarchaeota, such as acetate, can also serve as food or energy sources for Methanosaeta. Therefore, we speculate that these two groups prefer living in the >0.45 μm size fraction. Meanwhile, these processes of Bathyarchaeota and Methanosaeta not only could indicate the existence of a syntrophic association between these two groups, but also may explain the mechanism of symbiosis between Bathyarchaeota and Methanosaeta [56].
4.2. Microbial Candidate for In Situ Bioremediation in SGD
The in situ biodegradation or bioremediation of groundwater contamination has gradually gained more attention because it is more cost effective than commercial inoculum [59]. In this study, approximately 10% of bacterial sequences in both the >0.45 μm size fraction and the 0.2–0.45 μm fraction belonged to Novosphingobium spp. This Novosphingobium is a genus within the alpha subclass of Proteobacteria [60]; this subclass includes species of the genus Sphingomonas [61]. This bacterial group plays an important role in the degradation of aromatic compounds, such as PAHs, carbofuran, pentachlorophenol (PCP), and estrogen [60,62,63,64,65].
The dominant archaeal communities in the >0.45 μm size fraction in this study were Bathyarchaeota, which can also degrade aromatic compounds into small-molecule compounds, such as acetate [55]. Acetate is an important electron carrier, and may be consumed by heterotrophic bacteria and acetoclastic methanogens [55]. To further prove the accuracy of our results, the PICRUSt approach [32] was proposed to predict the abundance of functional categories (KEGG metabolic pathways) using 16S rRNA gene sequences in our study. The pathways involved in the degradation of aromatic compounds by both bacteria and archaea were identified and are shown in Table 5 and Table 6. Therefore, the high abundance of Bathyarchaeota in the >0.45 μm size fraction and Novosphingobium spp. in the >0.45 μm size fraction and the 0.2–0.45 μm fraction in our samples indicated that these species could be selected as microbial candidates for in situ biodegradation or bioremediation in polluted submarine groundwater.
As a significant component of iron cycling [66], Fe(II)-oxidizing bacteria (FeOB) of the family Gallionellaceae can obtain energy for growth by catalyzing the oxidation of Fe(II) [67] and then form Fe(III) mineral precipitation [34]. The sequences of OTU 40 showed 98.9% similarity with clone F22F60 from a Fe(II)-oxidizing, nitrate-reducing enrichment culture (FN430660) [37]. This process could cause Fe(II) oxidation and form Fe(III) mineral precipitation while consuming nitrate and producing N2 [37]. Because this process can reduce the concentration of nitrate in SGD, it plays a very important role in alleviating environmental problems caused by high concentrations of SGD-derived nitrate. The most abundant bacterial OTU, OTU 594, in the 0.2–0.45 μm fraction, was affiliated with Limnohabitans spp., which are characterized by high growth rates, metabolic flexibility and a preference for phytoplankton-derived organic material [68,69]. In addition, photolysis products of dissolved organic matter (DOM) have been suggested as an important source of substrates for Limnohabitans spp. [70,71,72]. Therefore, Gallionella spp. and Limnohabitans spp. in SGD could also be selected as microbial candidates for in situ biodegradation/bioremediation.
4.3. Influence of Key Microbes on SGD in Qinzhou Bay
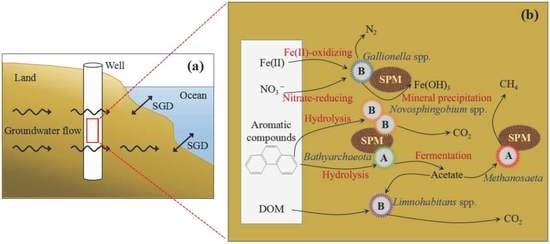
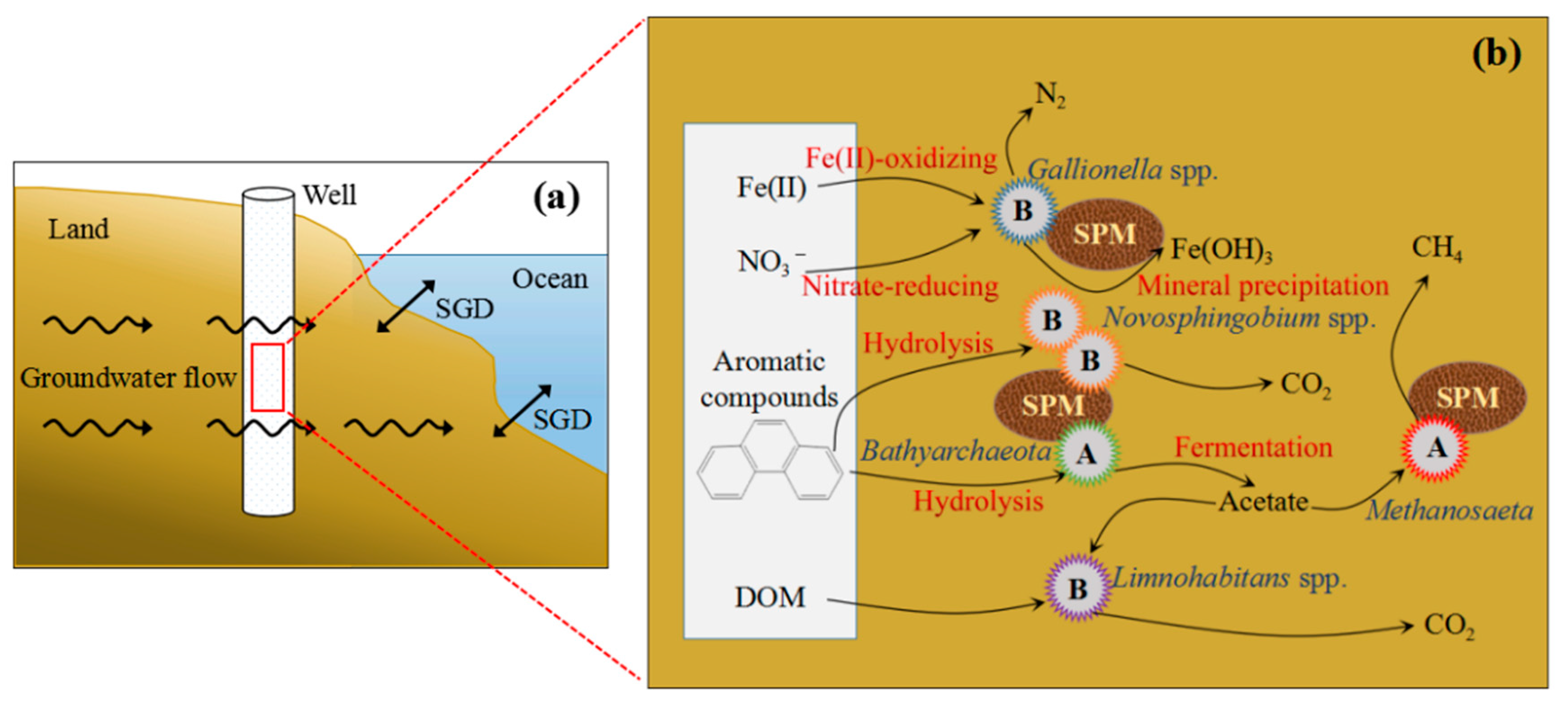
Generally, submarine groundwater systems contaminated by organic matter are electron-acceptor limited and are in reducing environments [9,73]. As a result of DOC oxidation, SGD may be depleted in NO3– and enriched in reduced metabolites such as dissolved Fe(II) [74,75]. Furthermore, aquifer systems often contain solid phase organic matter with minerals such as pyrite, siderite and Fe(II) silicates, which can reduce suitable oxidants such as NO3– or oxygen [76,77,78]. Excess SGD-derived nutrients, carbon and metals have been recognized to have deleterious effects on marine ecological environments. Microorganisms are recognized as important participants in biogeochemical processes in the SGD [22]. In Figure 7, Fe(II)-oxidizing and nitrate-reducing processes occur in FeOB in the >0.45 μm size fraction, and then generate Fe(III) mineral precipitation and N2, respectively [37,79]. These processes could reduce nitrate in SGD. Both Bathyarchaeota in the >0.45 μm size fraction and Novosphingobium spp. in the >0.45 μm size fraction and 0.2–0.45 μm fraction could degrade aromatic compounds [55,65]. The Methanosaeta and Limnohabitans spp. could consume acetate produced by Bathyarchaeota fermentation and then generate CH4 and CO2, respectively ([36,56,57]. Meanwhile, the Limnohabitans spp. could utilize the DOM as an important source of substrates [70,71,72]. Submarine groundwater often contains higher concentrations of nutrients, carbon, and metals than river water and seawater [3,9]. However, the concentrations of NO3– (5.2 μmol L−1) and DOC (1.0 mmol L−1) in our studied submarine groundwater were lower than those (NO3–: 7.2 μmol L−1; DOC: 1.1 mmol L−1) in the seawater of Qinzhou Bay. We suggest that the lower NO3– and DOC concentrations in our studied submarine groundwater are the result of the metabolic action of these bacteria and the archaea. In addition, the concentrations of DIN and DOC in global submarine groundwater were 211 μmol L−1 and 1.8 mmol L−1 [12,80], which significantly higher than those (DIN: 10.4 μmol L−1; DOC: 1.0 mmol L−1) in the submarine groundwater of Qinzhou Bay. This further indicates that nutrients and the carbon of submarine groundwater may be utilized by these microorganisms in our study site. In view of this, the dominant microbes of the two size fractions in the studied submarine groundwater of Qinzhou Bay were likely involved in the degradation or consumption of organic pollutants and excess nutrients, such as aromatic compounds, nitrates and DOM. These dominant microbes will be expected to reduce the amount of organic pollutants and excess nutrients from SGD entering coastal water, which may impact carbon, nitrogen and iron cycling in SGD (Figure 7).
5. Conclusions
In this study, we investigated the microbial assemblages from two size fractions in submarine groundwater near an industrial zone, Qinzhou Bay. The main bacterial and archaeal groups in the >0.45 μm size fraction and 0.2–0.45 μm fraction communities were significantly different, based on a PERMANOVA test. Moreover, some potential key microbial groups from two size fractions were also involved in the utilization and conversion of biogenic elements such as carbon, nutrients and iron. For example, Bathyarchaeota was detected in the >0.45 μm size fraction and Novosphingobium spp. were found in both the >0.45 μm size fraction and the 0.2–0.45 μm fraction, indicating that these genera play an important role in the degradation of aromatic compounds. Gallionella spp. were most abundant in the >0.45 μm size fraction and could consume nitrate and produce N2 while oxidizing Fe(II) to Fe(III) mineral precipitation, reflecting that this genus could consume a part of the nitrate in SGD before flowing into coastal water. These results showed that microbial communities may have potential roles in carbon, nitrogen and iron cycling in submarine groundwater systems (subterranean estuary).
In addition, the dominant bacterial sequences in the 0.2–0.45 μm fraction were affiliated with Limnohabitans spp., which could utilize DOM as an important source of substrates. Therefore, the Bathyarchaeota, Novosphingobium spp., Limnohabitans spp. and Gallionella spp. could be selected as microbial candidates for in situ biodegradation or bioremediation in the polluted subterranean estuary by aromatic compounds, excess nitrates and DOM. Both functional metagenomic analyses of the structure, function and culture experiments of isolating specific microbes for in situ bioremediation in the subterranean estuary, such as the aquifer along the coast of Qinzhou Bay (i.e., the polluted coastal zone by a petroleum refinery), are necessary to further any research in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4441/11/6/1261/s1, Table S1: DNA results for each sample.
Author Contributions
X.C. and Q.Y. designed the experiments; X.C. collected the samples; X.C. and Q.Y. performed the experiments and analyzed the data; X.C., Q.Y., J.D., and J.Z. wrote the manuscript.
Funding
This research was supported by the Natural Science Foundation of China (Grant No. 41576083).
Acknowledgments
We would like to thank all the colleagues of the biogeochemical group in the State Key Laboratory of Estuarine and Coastal Research at East China Normal University (SKLEC/ECNU) for their assistance in the field. We wish to thank the editors and two anonymous reviewers for their constructive comments for improvement of the original manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Burnett, W.C.; Bokuniewicz, H.; Huettel, M.; Moore, W.S.; Taniguchi, M. Groundwater and pore water inputs to the coastal zone. Biogeochemistry 2003, 66, 3–33. [Google Scholar] [CrossRef]
- Moore, W.S. The subterranean estuary: A reaction zone of ground water and sea water. Mar. Chem. 1999, 65, 111–125. [Google Scholar] [CrossRef]
- Moore, W.S. The effect of submarine groundwater discharge on the ocean. Annu. Rev. Mar. Sci. 2010, 2, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Pavlidou, A.; Papadopoulos, V.P.; Hatzianestis, I.; Simboura, N.; Patiris, D.; Tsabaris, C. Chemical inputs from a karstic submarine groundwater discharge (SGD) into an oligotrophic Mediterranean coastal area. Sci. Total Environ. 2014, 488, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Cukrov, N.; Du, J. Porewater-derived nutrient fluxes in a coastal aquifer (Shengsi Island, China) and its implication. Estuar. Coast. Shelf Sci. 2019, 218, 204–211. [Google Scholar] [CrossRef]
- Luo, X.; Jiao, J.J. Submarine groundwater discharge and nutrient loadings in Tolo Harbor, Hong Kong using multiple geotracer-based models, and their implications of red tide outbreaks. Water Res. 2016, 102, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Lee, Y.W.; Kim, G. Large submarine groundwater discharge and benthic eutrophication in Bangdu Bay on volcanic Jeju Island, Korea. Limnol. Oceanogr. 2005, 50, 1393–1403. [Google Scholar] [CrossRef]
- McCoy, C.; Viso, R.; Peterson, R.N.; Libes, S.; Lewis, B.; Ledoux, J.; Voulgaris, G.; Smith, E.; Sanger, D. Radon as an indicator of limited cross-shelf mixing of submarine groundwater discharge along an open ocean beach in the South Atlantic Bight during observed hypoxia. Cont. Shelf Res. 2011, 31, 1306–1317. [Google Scholar] [CrossRef]
- Slomp, C.P.; Van Cappellen, P. Nutrient inputs to the coastal ocean through submarine groundwater discharge: Controls and potential impact. J. Hydrol. 2004, 295, 64–86. [Google Scholar] [CrossRef]
- UNESCO. Submarine Groundwater Discharge: Management Implications, Measurements and Effects; IHP-VI, Series on Groundwater No. 5, IOC Manuals and Guides No. 44; UNESCO: Paris, France, 2004. [Google Scholar]
- Chen, X.; Lao, Y.; Wang, J.; Du, J.; Liang, M.; Yang, B. Submarine Groundwater-Borne Nutrients in a Tropical Bay (Maowei Sea, China) and Their Impacts on the Oyster Aquaculture. Geochem. Geophys. Geosyst. 2018, 19, 932–951. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, F.; Lao, Y.; Wang, X.; Du, J.; Santos, I.R. Submarine groundwater discharge-derived carbon fluxes in mangroves: An important component of blue carbon budgets? J. Geophys. Res. Oceans 2018, 123, 6962–6979. [Google Scholar] [CrossRef]
- Zhang, J.L.; Li, Y.Y.; Wang, Y.H.; Zhang, Y.Y.; Zhang, D.; Zhang, R.J.; Li, Y.; Zhang, G. Spatial distribution and ecological risk of polychlorinated biphenyls in sediments from Qinzhou Bay, Beibu Gulf of South China. Mar. Pollut. Bull. 2014, 80, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.G.; Lin, Q.; Yu, Z.L.; Wang, X.N.; Ke, C.L.; Ning, J.J. Speciation and risk of heavy metals in sediments and human health implications of heavy metals in edible nekton in Beibu Gulf, China: A case study of Qinzhou Bay. Mar. Pollut. Bull. 2015, 101, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Millward, G.E. Suspended particles: Their role in estuarine biogeochemical cycles. Estuar. Coast. Shelf Sci. 2002, 55, 857–883. [Google Scholar] [CrossRef]
- Suzumura, M.; Kokubun, H.; Arata, N. Distribution and characteristics of suspended particulate matter in a heavily eutrophic estuary, Tokyo Bay, Japan. Mar. Pollut. Bull. 2004, 49, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.; Bloesch, J.; Rathke, D.E. Sampling the settling and suspended particulate matter (SPM). In Handbook of Techniques for Aquatic Sediments Sampling; Murdoch, A., MacKnight, S.D., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 97–129. [Google Scholar]
- Uncles, R.J.; Joint, I.; Stephens, J.A. Transport and retention of suspended particulate matter and bacteria in the Humber-Ouse Estuary, United Kingdom, and their relationship to hypoxia and anoxia. Estuaries 1998, 21, 597–612. [Google Scholar] [CrossRef]
- Kravchishina, M.D.; Mitzkevich, I.N.; Veslopolova, E.F.; Shevchenko, V.P.; Lisitzin, A.P. Relationship between the suspended particulate matter and microorganisms in the White Sea waters. Oceanology 2008, 48, 837–854. [Google Scholar] [CrossRef]
- Perkins, T.L.; Perrow, K.; Rajko-Nenow, P.; Jago, C.F.; Jones, D.L.; Malham, S.K.; McDonald, J.E. Decay rates of faecal indicator bacteria from sewage and ovine faeces in brackish and freshwater microcosms with contrasting suspended particulate matter concentrations. Sci. Total Environ. 2016, 572, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Boehm, A.B.; Shellenbarger, G.G.; Paytan, A. Groundwater discharge: Potential association with fecal indicator bacteria in the surf zone. Environ. Sci. Technol. 2004, 38, 3558–3566. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, J.A.; Du, J.Z.; Zhang, J. Bacterial diversity in submarine groundwater along the coasts of the yellow sea. Front. Microbiol. 2016, 6, 1519. [Google Scholar] [CrossRef]
- Halliday, E.; Gast, R.J. Bacteria in beach sands: An emerging challenge in protecting coastal water quality and bather health. Environ. Sci. Technol. 2010, 45, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.M.; Hong, G.H.; Zhang, J.; Ye, X.W.; Jiang, X.L. Nutrient budgets for large Chinese estuaries. Biogeosciences 2009, 6, 2245–2263. [Google Scholar] [CrossRef] [Green Version]
- Cerdà-Domènech, M.; Rodellas, V.; Folch, A.; Garcia-Orellana, J. Constraining the temporal variations of Ra isotopes and Rn in the groundwater end-member: Implications for derived SGD estimates. Sci. Total Environ. 2017, 595, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, A.C.C.; Cleary, D.F.R.; Almeida, A.; Cunha, A.; Dealtry, S.; Mendonça-Hagler, L.C.S.; Smalla, K.; Gomes, N.C. Denaturing gradient gel electrophoresis and barcoded pyrosequencing reveal unprecedented archaeal diversity in mangrove sediment and rhizosphere samples. Appl. Environ. Microbiol. 2012, 78, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Li, Y.F.; Chen, Y.R.; Yang, J.L.; Bao, W.Y.; Guo, X.; Liang, X.; Shi, Z.Y.; Li, J.-L.; Ding, D.-W. Effects of substratum type on bacterial community structure in biofilms in relation to settlement of plantigrades of the mussel Mytilus coruscus. Int. Biodeter. Biodegr. 2014, 96, 41–49. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platformindependent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.R.; Thurber, R.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Cho, H.M.; Kim, G. Determining groundwater Ra end-member values for the estimation of the magnitude of submarine groundwater discharge using Ra isotope tracers. Geophys. Res. Lett. 2016, 43, 3865–3871. [Google Scholar] [CrossRef] [Green Version]
- Fabisch, M.; Beulig, F.; Akob, D.M.; Küsel, K. Surprising abundance of Gallionella-related iron oxidizers in creek sediments at pH 4.4 or at high heavy metal concentrations. Front. Microbiol. 2013, 4, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.W.; Kasalický, V.; Jezbera, J.; Brandt, U.; Šimek, K. Limnohabitans australis sp. nov., isolated from a freshwater pond, and emended description of the genus Limnohabitans. Int. J. Syst. Evol. Microbiol. 2010, 60, 2946–2950. [Google Scholar] [CrossRef] [PubMed]
- Kasalický, V.; Jezbera, J.; Šimek, K.; Hahn, M.W. Limnohabitans planktonicus sp. nov. and Limnohabitans parvus sp. nov., planktonic betaproteobacteria isolated from a freshwater reservoir, and emended description of the genus Limnohabitans. Int. J. Syst. Evol. Microbiol. 2010, 60, 2710–2714. [Google Scholar] [CrossRef]
- Blöthe, M.; Roden, E.E. Composition and activity of an autotrophic Fe (II)-oxidizing, nitrate-reducing enrichment culture. Appl. Environ. Microbiol. 2009, 75, 6937–6940. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.N.; Frigon, D.; Raskin, L. Populations related to Alkanindiges, a novel genus containing obligate alkane degraders, are implicated in biological foaming in activated sludge systems. Environ. Microbiol. 2007, 9, 1898–1912. [Google Scholar] [CrossRef]
- Sohn, J.H.; Kwon, K.K.; Kang, J.H.; Jung, H.B.; Kim, S.J. Novosphingobium pentaromativorans sp. nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. Int. J. Syst. Evol. Microbiol. 2004, 54, 1483–1487. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, M.S.; Jung, M.J.; Roh, S.W.; Shin, K.S.; Bae, J.W. Sphingopyxis soli sp. nov., isolated from landfill soil. Int. J. Syst. Evol. Microbiol. 2010, 60, 1682–1686. [Google Scholar] [CrossRef]
- Lee, S.; Weon, H.Y.; Kim, S.J.; Ahn, T.Y. Flavobacterium koreense sp. nov., Flavobacterium chungnamense sp. nov., and Flavobacterium cheonanense sp. nov., isolated from a freshwater reservoir. J. Microbiol. 2011, 49, 387–392. [Google Scholar] [CrossRef]
- LaFrentz, B.R.; Waldbieser, G.C.; Welch, T.J.; Shoemaker, C.A. Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J. Fish Dis. 2014, 37, 657–669. [Google Scholar] [CrossRef]
- Enright, A.M.; McGrath, V.; Gill, D.; Collins, G.; O’Flaherty, V. Effect of seed sludge and operation conditions on performance and archaeal community structure of low-temperature anaerobic solvent-degrading bioreactors. Syst. Appl. Microbiol. 2009, 32, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Takahata, Y.; Hoaki, T.; Watanabe, K. Physiological and molecular characterization of a microbial community established in unsaturated, petroleum-contaminated soil. Environ. Microbiol. 2005, 7, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Zhang, L.; Harnack, M.; Olson, M.V.; Kaul, R.; Ingram-Smith, C.; Smith, K.S. Complete genome sequence of Methanosaeta concilii, a specialist in aceticlastic methanogenesis. J. Bacteriol. 2011, 193, 3668–3669. [Google Scholar] [CrossRef] [PubMed]
- Llirós, M.; Casamayor, E.O.; Borrego, C. High archaeal richness in the water column of a freshwater sulfurous karstic lake along an interannual study. FEMS Microbiol. Ecol. 2008, 66, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, R.; Klose, M.; Noll, M.; Kemnitz, D.; Bodelier, P.L. Soil type links microbial colonization of rice roots to methane emission. Glob. Chang. Biol. 2008, 14, 657–669. [Google Scholar] [CrossRef]
- Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Hug, L.A.; Brown, C.T.; Wilkins, M.J.; Frischkorn, K.R.; Tringe, S.G.; Singh, A.; Markillie, L.M.; et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 2015, 25, 690–701. [Google Scholar] [CrossRef]
- Kubo, K.; Lloyd, K.G.; Biddle, J.F.; Amann, R.; Teske, A.; Knittel, K. Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J. 2012, 6, 1949–1965. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.G.; Schreiber, L.; Petersen, D.G.; Kjeldsen, K.U.; Lever, M.A.; Steen, A.D.; Stepanauskas, R.; Richter, M.; Kleindienst, S.; Lenk, S.; et al. Predominant archaea in marine sediments degrade detrital proteins. Nature 2013, 496, 215–218. [Google Scholar] [CrossRef]
- Evans, P.N.; Parks, D.H.; Chadwick, G.L.; Robbins, S.J.; Orphan, V.J.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C.S.; Baker, B.J.; Seitz, K.; Hyde, A.S.; Dick, G.J.; Hinrichs, K.U.; Teske, A.P. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 2016, 18, 1200–1211. [Google Scholar] [CrossRef]
- Fry, J.C.; Parkes, R.J.; Cragg, B.A.; Weightman, A.J.; Webster, G. Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 2008, 66, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Xu, J.; Qin, D.; He, Y.; Xiao, X.; Wang, F. Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 2014, 8, 650–659. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, M.; Perumal, V.; Feng, X.; Fang, J.; Xie, J.; Sievert, S.M.; Wang, F. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 2016, 1, 16035. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, R.; Wang, H.; Gong, L.; Man, B.; Xu, Y. Distribution of Bathyarchaeota Communities Across Different Terrestrial Settings and Their Potential Ecological Functions. Sci. Rep. 2017, 7, 45028. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Liu, X.; Dong, X. Methanosaeta harundinacea sp. nov., a novel acetate-scavenging methanogen isolated from a UASB reactor. Int. J. Syst. Evol. Microbiol. 2006, 56, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; He, M.; Yang, Z.; Lin, C.; Quan, X.; Wang, H. Distribution of polycyclic aromatic hydrocarbons in water, suspended particulate matter and sediment from Daliao River watershed, China. Chemosphere 2007, 68, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Wu, W.; Gentry, T.J.; Carley, J.; Corbin, G.A.; Carroll, S.L.; Watson, D.B.; Jardine, P.M.; Zhou, J.; Criddle, C.S.; et al. Bacterial community succession during in situ uranium bioremediation: Spatial similarities along controlled flow paths. ISME J. 2009, 3, 47–64. [Google Scholar] [CrossRef]
- Gan, H.M.; Hudson, A.O.; Rahman, A.Y.A.; Chan, K.G.; Savka, M.A. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: Insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genom. 2013, 14, 431. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Yano, I.; Oyaizu, H.; Hashimoto, Y.; Ezaki, T.; Yamamoto, H. Proposals of Sphingomonas paucimobilis gen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov., Sphingomonas yanoikuyae sp. nov., Sphingomonas adhaesiva sp. nov., Sphingomonas capsulata comb. nov., and two genospecies of the genus Sphingomonas. Microbiol. Immunol. 1990, 34, 99–119. [Google Scholar] [CrossRef]
- Tiirola, M.A.; Männistö, M.K.; Puhakka, J.A.; Kulomaa, M.S. Isolation and characterization of Novosphingobium sp. strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl. Environ. Microbiol. 2002, 68, 173–180. [Google Scholar] [CrossRef]
- Yan, Q.X.; Hong, Q.; Han, P.; Dong, X.J.; Shen, Y.J.; Li, S.P. Isolation and characterization of a carbofuran-degrading strain Novosphingobium sp. FND-3. FEMS Microbiol. Lett. 2007, 271, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lai, Q.; Zheng, T.; Shao, Z. Novosphingobium indicum sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from a deep-sea environment. Int. J. Syst. Evol. Microbiol. 2009, 59, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Onda, K.; Morita, T.; Luxmy, B.S.; Tada, K.; Miya, A.; Murakami, T. Contribution of the estrogen-degrading bacterium Novosphingobium sp. strain JEM-1 to estrogen removal in wastewater treatment. J. Environ. Eng. 2010, 136, 890–896. [Google Scholar] [CrossRef]
- Kappler, A.; Straub, K.L. Geomicrobiological cycling of iron. Rev. Mineral. Geochem. 2005, 59, 85–108. [Google Scholar] [CrossRef]
- Konhauser, K.O.; Amskold, L.; Lalonde, S.V.; Posth, N.R.; Kappler, A.; Anbar, A. Decoupling photochemical Fe (II) oxidation from shallow-water BIF deposition. Earth Planet. Sci. Lett. 2007, 258, 87–100. [Google Scholar] [CrossRef]
- Pérez, M.T.; Sommaruga, R. Differential effect of algal-and soil-derived dissolved organic matter on alpine lake bacterial community composition and activity. Limnol. Oceanogr. 2006, 51, 2527–2537. [Google Scholar] [CrossRef]
- Šimek, K.; Kasalický, V.; Zapomělová, E.; Horňák, K. Alga-derived substrates select for distinct betaproteobacterial lineages and contribute to niche separation in Limnohabitans strains. Appl. Environ. Microbiol. 2011, 77, 7307–7315. [Google Scholar] [CrossRef]
- Hörtnagl, P.; Pérez, M.T.; Zeder, M.; Sommaruga, R. The bacterial community composition of the surface microlayer in a high mountain lake. FEMS Microbiol. Ecol. 2010, 73, 458–467. [Google Scholar] [CrossRef]
- Glaeser, S.P.; Grossart, H.P.; Glaeser, J. Singlet oxygen, a neglected but important environmental factor: Short-term and long-term effects on bacterioplankton composition in a humic lake. Environ. Microbiol. 2010, 12, 3124–3136. [Google Scholar] [CrossRef]
- Kasalický, V.; Jezbera, J.; Hahn, M.W.; Šimek, K. The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS ONE 2013, 8, e58209. [Google Scholar] [CrossRef]
- Chapelle, F. Ground-Water Microbiology and Geochemistry; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Chapelle, F.H.; Lovley, D.R. Competitive exclusion of sulfate reduction by Fe (lll)-reducing bacteria: A mechanism for producing discrete zones of high-iron ground water. Groundwater 1992, 30, 29–36. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; Balkema: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Postma, D.; Boesen, C.; Kristiansen, H.; Larsen, F. Nitrate reduction in an unconfined sandy aquifer: Water chemistry, reduction processes, and geochemical modeling. Water Resour. Res. 1991, 27, 2027–2045. [Google Scholar] [CrossRef]
- Engesgaard, P.; Kipp, K.L. A geochemical transport model for redox-controlled movement of mineral fronts in groundwater flow systems: A case of nitrate removal by oxidation of pyrite. Water Resour. Res. 1992, 28, 2829–2843. [Google Scholar] [CrossRef]
- Hartog, N.; Griffioen, J.; van der Weijden, C.H. Distribution and reactivity of O2-reducing components in sediments from a layered aquifer. Environ. Sci. Technol. 2002, 36, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Straub, K.L.; Benz, M.; Schink, B.; Widdel, F. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol. 1996, 62, 1458–1460. [Google Scholar] [PubMed]
- Cho, H.M.; Kim, G.; Kwon, E.Y.; Moosdorf, N.; Garcia-Orellana, J.; Santos, I.R. Radium tracing nutrient inputs through submarine groundwater discharge in the global ocean. Sci. Rep. 2018, 8, 2439. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Map showing the study site location (a), sampling site and industrial zone (b) in Qinzhou Bay.
Figure 1.
Map showing the study site location (a), sampling site and industrial zone (b) in Qinzhou Bay.

Figure 2.
The principal-coordinate analyses (PCoA) analysis on operational taxonomic unit (OTU) levels of main bacterial (a) and archaeal (b) genera from >0.45 μm (green dots) and 0.2–0.45 μm (red squares) size fractions. BP and BF represent bacteria of >0.45 μm and 0.2–0.45 μm size fractions, respectively. AP and AF represent archaea of >0.45 μm and 0.2–0.45 μm size fractions, respectively. The values of 1–4 represent four parallel samples.
Figure 2.
The principal-coordinate analyses (PCoA) analysis on operational taxonomic unit (OTU) levels of main bacterial (a) and archaeal (b) genera from >0.45 μm (green dots) and 0.2–0.45 μm (red squares) size fractions. BP and BF represent bacteria of >0.45 μm and 0.2–0.45 μm size fractions, respectively. AP and AF represent archaea of >0.45 μm and 0.2–0.45 μm size fractions, respectively. The values of 1–4 represent four parallel samples.

Figure 3.
Distributions of genus-level taxa for bacterial (a) and archaeal (b) samples. Bars represent the proportion of sequences represented by each genus. Bacterial taxa represented by less than 2% reads are pooled as “others”. The designation “norank” means that the specific archaeal taxa cannot be classified at the genus level. The proportion of each sequence in two size fractions is the mean value of quadruplicate analyses. BP, BF, AP and AF represent the average values of BP1–BP4, BF1–BF4, AP1–AP4 and AF1–AF4, respectively.
Figure 3.
Distributions of genus-level taxa for bacterial (a) and archaeal (b) samples. Bars represent the proportion of sequences represented by each genus. Bacterial taxa represented by less than 2% reads are pooled as “others”. The designation “norank” means that the specific archaeal taxa cannot be classified at the genus level. The proportion of each sequence in two size fractions is the mean value of quadruplicate analyses. BP, BF, AP and AF represent the average values of BP1–BP4, BF1–BF4, AP1–AP4 and AF1–AF4, respectively.

Figure 4.
Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship among the representative bacterial OTUs obtained in the >0.45 μm size fraction and the 0.2–0.45 μm fraction and reference 16S rRNA gene sequences retrieved from the NCBI GenBank. These bacterial OTUs were represented by the top 10 reads from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown. Bold bacterial OTUs were obtained from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The numbers in parentheses indicate the percentage composition of reads in each sample in the following order: (BP, BF). Methanococcus maripaludis (U38941) was used as the outgroup. The number in parentheses represents the mean value of quadruplicate analyses.
Figure 4.
Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship among the representative bacterial OTUs obtained in the >0.45 μm size fraction and the 0.2–0.45 μm fraction and reference 16S rRNA gene sequences retrieved from the NCBI GenBank. These bacterial OTUs were represented by the top 10 reads from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown. Bold bacterial OTUs were obtained from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The numbers in parentheses indicate the percentage composition of reads in each sample in the following order: (BP, BF). Methanococcus maripaludis (U38941) was used as the outgroup. The number in parentheses represents the mean value of quadruplicate analyses.

Figure 5.
Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship among the representative archaeal OTUs obtained in the >0.45 μm size fraction and the 0.2–0.45 μm fraction and reference 16S rRNA gene sequences retrieved from the NCBI GenBank. These archaeal OTUs were represented by the top 10 reads from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown. Bold archaeal OTUs were obtained from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The numbers in parentheses indicate the percentage composition of reads in each sample in the following order: (AP, AF). Limnohabitans australis strain MWH-BRAZ-DAM2D (NR_125544) was used as the outgroup. The number in parentheses represents the mean value of quadruplicate analyses.
Figure 5.
Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences showing the phylogenetic relationship among the representative archaeal OTUs obtained in the >0.45 μm size fraction and the 0.2–0.45 μm fraction and reference 16S rRNA gene sequences retrieved from the NCBI GenBank. These archaeal OTUs were represented by the top 10 reads from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown. Bold archaeal OTUs were obtained from the >0.45 μm size fraction and the 0.2–0.45 μm fraction. The numbers in parentheses indicate the percentage composition of reads in each sample in the following order: (AP, AF). Limnohabitans australis strain MWH-BRAZ-DAM2D (NR_125544) was used as the outgroup. The number in parentheses represents the mean value of quadruplicate analyses.

Figure 6.
Neighbor-joining phylogenetic tree showing the placement of three representative bathyarchaeotal OTUs in the study in the Bathyarchaeota subgroup 6. The reference sequences of different Bathyarchaeota subgroups are according to [52]. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown.
Figure 6.
Neighbor-joining phylogenetic tree showing the placement of three representative bathyarchaeotal OTUs in the study in the Bathyarchaeota subgroup 6. The reference sequences of different Bathyarchaeota subgroups are according to [52]. The scale bar represents the estimated number of nucleotide changes per sequence position. Percentage numbers on nodes refer to 1000 bootstrap resamplings conducted. Only values >50% are shown.

Figure 7.
Schematic diagram of submarine groundwater discharge (SGD) (a). Main mechanism of nitrate, carbon and iron depletion contributed by the dominant microbial community of SGD in Qinzhou Bay (b). The “Suspended particulate matter (SPM)” in ovals represents suspended particulate matter. The “A” and “B” in the star with different colors represent the specific archaeal and bacterial groups, respectively.
Figure 7.
Schematic diagram of submarine groundwater discharge (SGD) (a). Main mechanism of nitrate, carbon and iron depletion contributed by the dominant microbial community of SGD in Qinzhou Bay (b). The “Suspended particulate matter (SPM)” in ovals represents suspended particulate matter. The “A” and “B” in the star with different colors represent the specific archaeal and bacterial groups, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Descriptions of the sampling site and environmental chemical parameters of submarine groundwater.
Table 1.
Descriptions of the sampling site and environmental chemical parameters of submarine groundwater.
| Site Descriptions | Environmental Chemical Parameters | ||
|---|---|---|---|
| Latitude | 21°43′39.92″ N | Temperature (°C) | 17.2 |
| Longitude | 108°37′30.43″ E | Salinity | 0.5 |
| Location | Qinzhou City, Guangxi Province | 222Rn (Bq m−3) | 4140 |
| Well depth (m) | ~ 4 | DIC (mmol L−1) | 1.3 |
| Characteristics of sampling site | In the Gaoshatou Village; near an industrial zone; 800 meters from the coast; garbage found | DOC (mmol L−1) | 1.0 |
| NO3− (μmol L−1) | 5.2 | ||
| NO2− (μmol L−1) | 0.42 | ||
| Characteristics of submarine groundwater | Turbid water; smelly water | NH4+ (μmol L−1) | 4.8 |
| PO43− (μmol L−1) | 0.31 | ||
| SiO32− (μmol L−1) | 166 | ||
Table 2.
Calculations of Statistic R2 and p values using the PERmutational Multivariate ANalysis of VAriance (PERMANOVA) approach.
Table 2.
Calculations of Statistic R2 and p values using the PERmutational Multivariate ANalysis of VAriance (PERMANOVA) approach.
| Method | Distance | Bacteria | Archaea | ||
|---|---|---|---|---|---|
| R2 | p Value | R2 | p Value | ||
| PERMANOVA | Bray-Curtis | 0.51 | 0.02 | 0.78 | 0.02 |
Table 3.
Closest relatives of main OTUs in bacterial samples.
| OTU | Proportion of OTUs (%) | Closest Relatives | Similarity | |
|---|---|---|---|---|
| BP | BF | |||
| OTU 594 | 4.7 | 21.8 | Limnohabitans australis strain MWH-BRAZ-DAM2D [35] | NR_125544 (99.2%) |
| OTU 571 | 1.2 | 2.6 | Limnohabitans parvus strain II-B4 [36] | NR_125542 (98.9%) |
| OTU 40 | 16.6 | 0.2 | Clone F22F60 from a Fe(II)-oxidizing, nitrate-reducing enrichment culture [37] | FN430660 (98.9%) |
| OTU 1039 | 4.5 | 0.8 | Gammaproteobacterial clone SWL18 from a sludge wastewater treatment system [38] | AY528817 (99.7%) |
| OTU 261 | 10.7 | 5.4 | Novosphingobium pentaromativorans strain US6-1 [39] | NR_025248 (98.4%) |
| OTU 555 | 1.2 | 1.5 | Sphingopyxis soli strain BL03 [40] | NR_116739 (99.2%) |
| OTU 843 | 3.1 | 2.7 | Flavobacterium chungnamense strain ARSA-103 [41] | NR_117494 (98.7%) |
| OTU 591 | 0.1 | 1.6 | Flavobacterium columnare strain ATCC 23463 [42] | NR_118582 (98.4%) |
Table 4.
Closest relatives of main OTUs in archaeal samples.
| OTU | Proportion of OTUs (%) | Closest Relatives | Similarity | |
|---|---|---|---|---|
| AP | AF | |||
| OTU 563 | 29.4 (OTU 563, 516 and 598) | 3.0 (OTU 563, 516 and 598) | Clone R5ENDA4 from low-temperature anaerobic solvent-degrading bioreactors [43] | DQ399807 (100%) |
| OTU 516 | Clone ASC27 in unsaturated, petroleum-contaminated soil [44] | AB161330 (100%) | ||
| OTU 598 | Clone ASC37 in unsaturated, petroleum-contaminated soil [44] | AB161337 (100%) | ||
| OTU 540 | 19.5 | 3.3 | Acetoclastic methanoarchaeon Methanosaeta concilii strain GP6 [45] | NR_102903 (99.6%) |
| OTU 312 | 1.1 | 3.7 | DGGE gel band VIARC-174 from the water column of Lake Vilar [46] | EU683427 (99.2%) |
| OTU 458 | 3.2 | 7.4 | Clone WsL03b13_109f from groundwater of a volcanic mountain, Mt. Fuji (Unpublished) | AB794582 (98.4%) |
| OTU 536 | 1.8 | 5.7 | Clone LL_ADT_15 from low-level bank soil of the River Rhine [47] | AM503273 (98.8%) |
| OTU 541 | 0.5 | 2.3 | Clone gwa2_scaffold_29719 from an aquifer adjacent to Colorado River [48] | KP308734 (87.0%) |
| OTU 449 | 16.3 | 41.4 | VIARC-45 retrieved from the water column of Lake Vilar [46] | AM697998 (92.5%) |
Table 5.
Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways involved in the degradation of aromatic compounds in bacterial samples.
Table 5.
Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways involved in the degradation of aromatic compounds in bacterial samples.
| Pathway | Relative Abundance | Definition | |
|---|---|---|---|
| >0.45 μm Size Fraction | 0.2–0.45 μm Fraction | ||
| ko00351 | 0.00 | 0.00 | 1,1,1-Trichloro-2,2-bis(4-chlorophenyl)ethane (DDT) degradation |
| ko00361 | 0.22 | 0.21 | Chlorocyclohexane and chlorobenzene degradation |
| ko00362 | 1.31 | 1.55 | Benzoate degradation |
| ko00363 | 0.19 | 0.23 | Bisphenol degradation |
| ko00364 | 0.13 | 0.12 | Fluorobenzoate degradation |
| ko00621 | 0.21 | 0.18 | Dioxin degradation |
| ko00622 | 0.20 | 0.15 | Xylene degradation |
| ko00623 | 0.34 | 0.34 | Toluene degradation |
| ko00624 | 0.27 | 0.29 | Polycyclic aromatic hydrocarbon degradation |
| ko00625 | 0.50 | 0.52 | Chloroalkane and chloroalkene degradation |
| ko00626 | 0.48 | 0.54 | Naphthalene degradation |
| ko00627 | 0.88 | 1.09 | Aminobenzoate degradation |
| ko00633 | 0.16 | 0.13 | Nitrotoluene degradation |
| ko00642 | 0.22 | 0.18 | Ethylbenzene degradation |
| ko00643 | 0.20 | 0.20 | Styrene degradation |
| ko00791 | 0.10 | 0.10 | Atrazine degradation |
| ko00903 | 0.76 | 0.92 | Limonene and pinene degradation |
| ko00930 | 0.56 | 0.70 | Caprolactam degradation |
| Total | 6.73 | 7.46 | |
Table 6.
KEGG metabolic pathways involved in the degradation of aromatic compounds in archaeal samples.
Table 6.
KEGG metabolic pathways involved in the degradation of aromatic compounds in archaeal samples.
| Pathway | Relative Abundance | Definition | |
|---|---|---|---|
| >0.45 μm Size Fraction | 0.2–0.45 μm Fraction | ||
| ko00281 | 0.19 | 0.25 | Geraniol degradation |
| ko00361 | 0.01 | 0.03 | Chlorocyclohexane and chlorobenzene degradation |
| ko00362 | 0.49 | 0.53 | Benzoate degradation |
| ko00363 | 0.01 | 0.04 | Bisphenol degradation |
| ko00364 | 0.00 | 0.01 | Fluorobenzoate degradation |
| ko00621 | 0.01 | 0.03 | Dioxin degradation |
| ko00622 | 0.00 | 0.01 | Xylene degradation |
| ko00623 | 0.33 | 0.29 | Toluene degradation |
| ko00624 | 0.11 | 0.12 | Polycyclic aromatic hydrocarbon degradation |
| ko00625 | 0.17 | 0.23 | Chloroalkane and chloroalkene degradation |
| ko00626 | 0.05 | 0.11 | Naphthalene degradation |
| ko00627 | 0.23 | 0.29 | Aminobenzoate degradation |
| ko00630 | 1.03 | 1.05 | Glyoxylate and dicarboxylate metabolism |
| ko00633 | 0.87 | 0.77 | Nitrotoluene degradation |
| ko00642 | 0.01 | 0.02 | Ethylbenzene degradation |
| ko00643 | 0.01 | 0.03 | Styrene degradation |
| ko00791 | 0.00 | 0.02 | Atrazine degradation |
| ko00903 | 0.27 | 0.29 | Limonene and pinene degradation |
| ko00930 | 0.18 | 0.20 | Caprolactam degradation |
| Total | 3.97 | 4.31 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, X.; Ye, Q.; Du, J.; Zhang, J. Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone. Water 2019, 11, 1261. https://doi.org/10.3390/w11061261
AMA Style
Chen X, Ye Q, Du J, Zhang J. Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone. Water. 2019; 11(6):1261. https://doi.org/10.3390/w11061261
Chicago/Turabian StyleChen, Xiaogang, Qi Ye, Jinzhou Du, and Jing Zhang. 2019. "Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone" Water 11, no. 6: 1261. https://doi.org/10.3390/w11061261
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.